Elizabeth Chen ’28
Since the 2017 FDA approval of tisagenlecleucel, commonly known as Kymriah, for the treatment of patients under the age of 25 with relapsed or refractory acute lymphocytic leukemia, research in the field of CAR-T cell therapy has continued to rapidly develop.
CAR-T cell therapy is a gene therapy using CAR-T cells: T-cells genetically engineered to artificially express a chimeric antigen receptor (CAR). Autologous CAR-T cell therapy uses an individual’s T-cells, rather than those of a donor, to precisely target cancerous cells in the body. Following leukapheresis (collection of a patient’s white blood cells) and the separation of T cells from the white blood cells, a gene coding for a chimeric antigen receptor protein is inserted into the T-cells, allowing them to express the chimeric antigen receptor on the cell surface. After millions of cells are grown and multiplied, they are infused back into the patient. This leads to significant advantages in treatment time and remission length as CAR-T cells can persist in the body in the long term in the correct conditions (Labanieh et al., 2018).
There are currently six FDA-approved CAR-T cell therapies. In almost all cases, CAR-T cell therapy is only viable when multiple other treatment options have already been used and failed to control the cancer. Once a third-line treatment, CAR-T cell therapy is now recognized as an effective second-line treatment for certain cancers, including relapsed or refractory large B-cell lymphoma (Kamdar et al., 2022), due to improved safety and strong clinical outcomes.
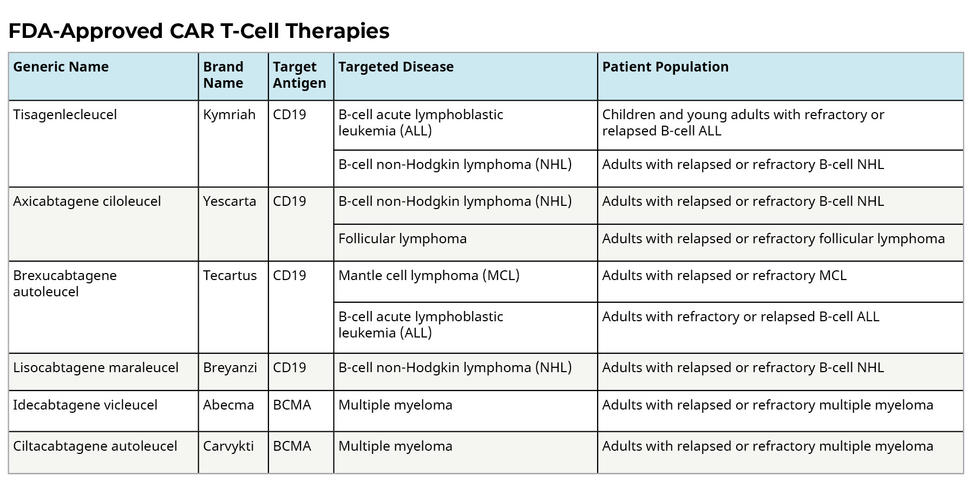
Six FDA-approved CAR-T cell therapies (NIH, 2022).
Class II major histocompatibility complex (MHC II) proteins allow helper T-cells to recognize foreign pathogens by displaying peptide fragments from extracellular proteins, specifically pathogens, on the surface of somatic cells (Labanieh et al., 2018). However, cancerous cells downregulate MHC II proteins and other factors important in antigen presentation. Therefore, T-cells are largely unable to recognize cancerous cells in the body and trigger an immune response (Sterner & Sterner, 2021). CAR-T cells artificially express a CAR, allowing them to directly recognize and attach to targeted tumor-associated antigens (TAAs) which exist on all cancerous cells but are absent or found in lower quantities on healthy somatic cells (Labanieh et al., 2018). Because CARs can recognize TAAs in an MHC-unrestricted manner, they can independently target and eradicate cancerous cells in the body (Sterner & Sterner, 2021). Currently, TAAs for FDA-approved CAR-T cell therapies include CD19 and BCMA.
The most significant challenge currently faced by researchers is the potential for relapse after CAR-T cell therapy, defined by the persistence of tumor cells after achieving complete remission. Relapse after CAR-T cell therapy was observed in 40-60% of patients with B-cell malignancies and 30-60% of patients with relapsed/refractory ALL (Wang & Wang, 2023). This is often due to several reasons. One explanation is antigen escape, which occurs with the total or partial loss of the target antigen recognized by the CAR (Negishi et al., 2024). Tumor cells with low or negative antigen expression levels cannot be targeted by CAR-T cells and may undergo clonal expansion, resulting in relapse. Another explanation is the immunosuppressive tumor microenvironment, in which interactions with various cells can cause CAR-T cells to become exhausted in vivo and have decreased persistence (Negishi et al., 2024). Interactions between programmed cell death protein-1 (PD-1) on tumor-specific T cells with programmed cell death-ligand 1 (PD-L1) on tumor-associated macrophages, myeloid-derived suppressor cells, or cancer-associated fibroblasts (CAFs) found in the TME cause cell death and reduce the number of CAR-T cells present following treatment. Finally, CAR-T cell exhaustion is associated with reduced therapeutic efficacy and resistance to treatment (Negishi et al., 2024). Specifically, the existence of a large quantity of exhausted CD8+ T cells is associated with relapsed and refractory outcomes.
The recent development of a CAR enhancer (CAR-E) platform at the Dana Farber Cancer Institute addresses these obstacles in a novel way. The CAR-E platform is made by combining an antigen normally targeted by CAR-T cells, such as BCMA, which was used in this work, and a low-affinity enhancer molecule to allow CAR-T cells to allow for more precise targeting and to facilitate the delivery of immune-modulating agents after infusion (Rakhshandehroo et al., 2024). Specifically, the interleukin-2 (IL-2) cytokine, when fused to a tumor-associated antigen, promotes T-cell activation and enhances antitumor activity, selectively inducing IL-2 signaling upon CAR-antigen binding. Engagement of both the IL-2 signaling pathway and CAR endodomain signaling pathway is necessary for CAR-E function: treatment with nontargeted low-affinity IL-2 does not yield as potent effects as observed in treatment with BCMA-mutIL2 (i.e. targeted low-affinity IL-2), while the absence of a CAR endodomain causes CAR-E to lose its effect on CAR-T cells as the presence of the tumor-associated antigen alone activates CAR-T cells at high concentrations in vitro but cannot expand CAR-T cells in vivo (Rakhshandehroo et al., 2024).
Thus, the CAR-E platform addresses two significant obstacles. First, by increasing CAR-T cell proliferation and clearance of tumor cells (Rakhshandehroo et al., 2024), CAR-E enables CAR-T cells to persist, unexhausted, through treatment and following remission. Secondly, CAR-E enhances the development of a wide range of memory CAR-T cells, including stem cell-like memory T cells, central memory T cells, and effector memory T cells, enhancing immune response following remission and decreasing the potential for and severity of relapse.
Using an MM xenograft mouse model, Rakhshandehroo et al. (2024) showed that the BCMA-mutIL-2 CAR-E led to enhanced persistence of CAR-T cells in vivo, and as well that CAR-E associated CAR-T cells function even 3 months after infusion in a mouse model. Furthermore, it was demonstrated that mice treated with a low dose of BCMA-mutIL-2 CAR-E of only 100,000 CAR-T cells achieved complete tumor clearance. This provided evidence for a crucial therapeutic and practical benefit of CAR-E: it allows patients to be treated with a lower dose of CAR-T cells, allowing the side effects of cytokine release syndrome (CRS), an aggressive immune response resulting in fever, nausea, rapid heartbeat, and potential neurological problems that must be managed carefully following treatment, associated with the current clinical approach of infusing hundreds of millions of CAR-T cells during treatment, to be mitigated. Alongside its ability to reduce the number of CAR-T cells needed for treatment, CAR-E may also reduce or eliminate the need for lymphodepleting chemotherapy before treatment, which results in an increased concentration of T-cell stimulating cytokines (including IL-7 and IL-15) and increases the risk for potent side effects associated with CRS. The deliverance of IL-2 to CAR-T cells is similar to IL-15 as they use the same signaling receptors, such that selectively delivering IL-2 to CAR-T cells could remove the need for lymphodepletion.
Going forward, CAR-E has the potential to be adapted to CAR-T cells in other liquid tumors with different target antigens, including CD19 and CD22, as well as in solid tumors and in the developing research of CAR-NK cells. This study offers a unique solution to existing challenges in CAR-T cell therapy while contributing to advances in the investigation of CAR-T cell therapy as a standalone treatment for hematological malignancies.
Elizabeth Chen is a staff writer at The Princeton Medical Review. She can be reached at ec8469@princeton.edu.
References
Kamdar, M., Solomon, S. R., Arnason, J., Johnston, P. B., Glass, B., Bachanova, V., Ibrahimi, S.,
Mielke, S., Mutsaers, P., Hernandez-Ilizaliturri, F., Izutsu, K., Morschhauser, F., Lunning, M., Maloney, D. G., Crotta, A., Montheard, S., Previtali, A., Stepan, L., Ogasawara, K., … Abramson, J. S. (2022). Lisocabtagene Maraleucel versus standard of care with salvage chemotherapy followed by autologous stem cell transplantation as second-line treatment in patients with relapsed or refractory large B-cell lymphoma (transform): Results from an interim analysis of an open-label, randomised, phase 3 trial. The Lancet, 399(10343), 2294–2308. https://doi.org/10.1016/s0140-6736(22)00662-6
Labanieh, L., Majzner, R.G., & Mackall, C.L. (2018). Programming CAR-T cells to kill cancer.
Nat. Biomed. Eng., 2(6), 377–391. https://doi.org/10.1038/s41551-018-0235-9
National Cancer Institute. (2022, March 10). CAR T Cells: Engineering Patients’ Immune Cells
to Treat Their Cancers. https://www.cancer.gov/about-cancer/treatment/research/car-t-cells
Negishi, S., Girsch, J. H., Siegler, E. L., Bezerra, E. D., Miyao, K., & Sakemura, R. L. (2024).
Treatment strategies for relapse after car T-cell therapy in B cell lymphoma. Frontiers in Pediatrics, 11. https://doi.org/10.3389/fped.2023.1305657
Rakhshandehroo, T., Mantri, S.R., Moravej, H. et al. (2024). A CAR enhancer increases the
activity and persistence of CAR T cells. Nat Biotechnol.
https://doi.org/10.1038/s41587-024-02339-4
Sterner, R.C., & Sterner, R.M. (2021). CAR-T cell therapy: current limitations and potential
strategies. Blood Cancer J., 11(4), 69. https://doi.org/10.1038/s41408-021-00459-7
Wang, J.Y. & Wang L. (2023). CAR-T cell therapy: Where are we now, and where are we heading? Blood Sci., 5(4), 237-248. https://doi.org/10.1097/BS9.0000000000000173.
Leave a Reply